We explore the chemical world by modeling large molecular systems on the computer
and looking at their diverse properties such as structure, energies, dynamics and spectroscopic quantities.
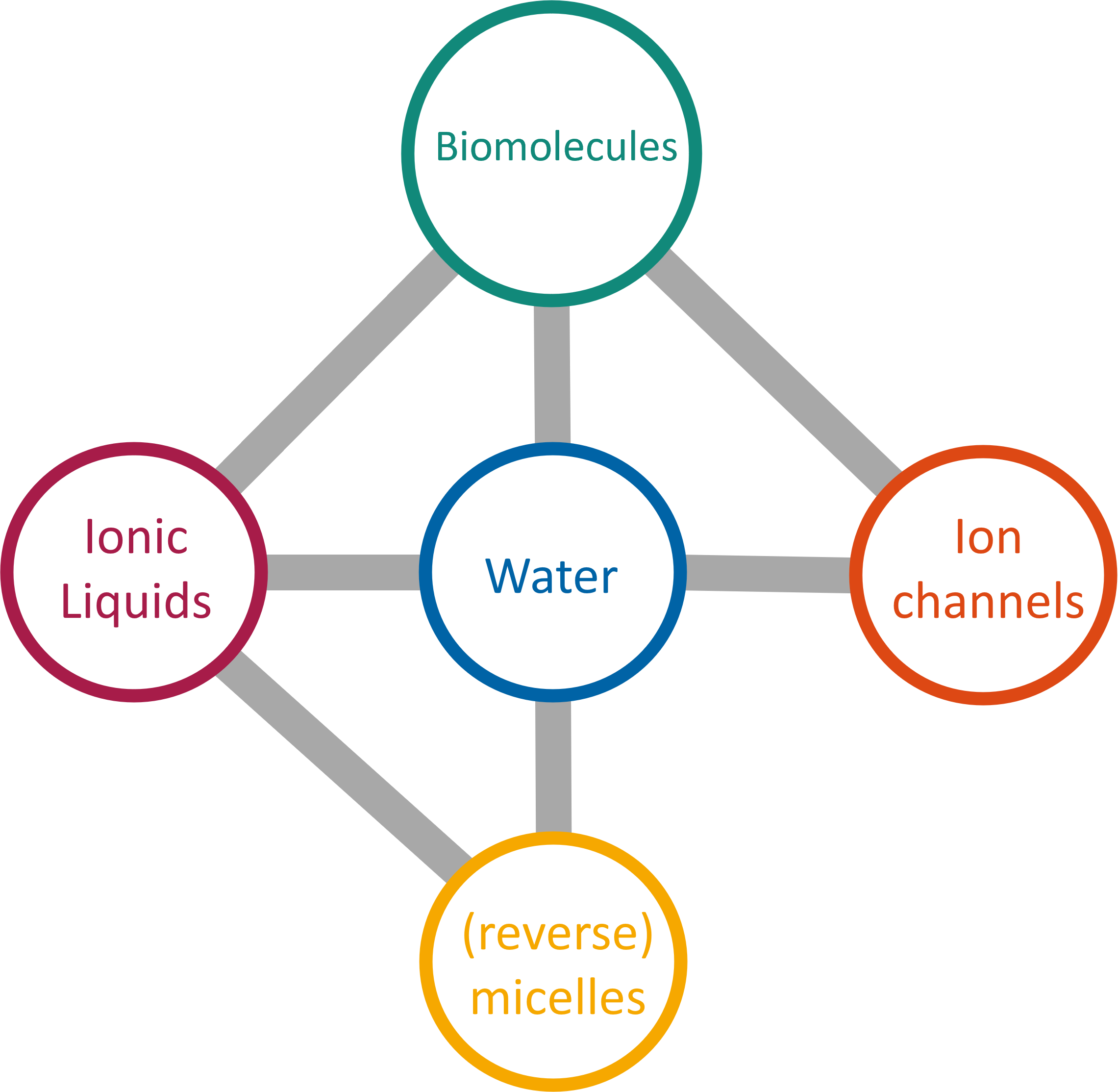
We target systems that include, but are not limited to, proteins in solvated environments or complex electrolytes such as ionic liquids.
Our investigations also encompass the study of micellar structures, both conventional and reversed, to elucidate their effects on
the molecules located within the core and at the interfaces of these nanoscale entities.
These systems, often comprising thousands of atoms, are studied to understand their behaviors over extended time scales of
several hundred nanoseconds, relevant to experimental observables.
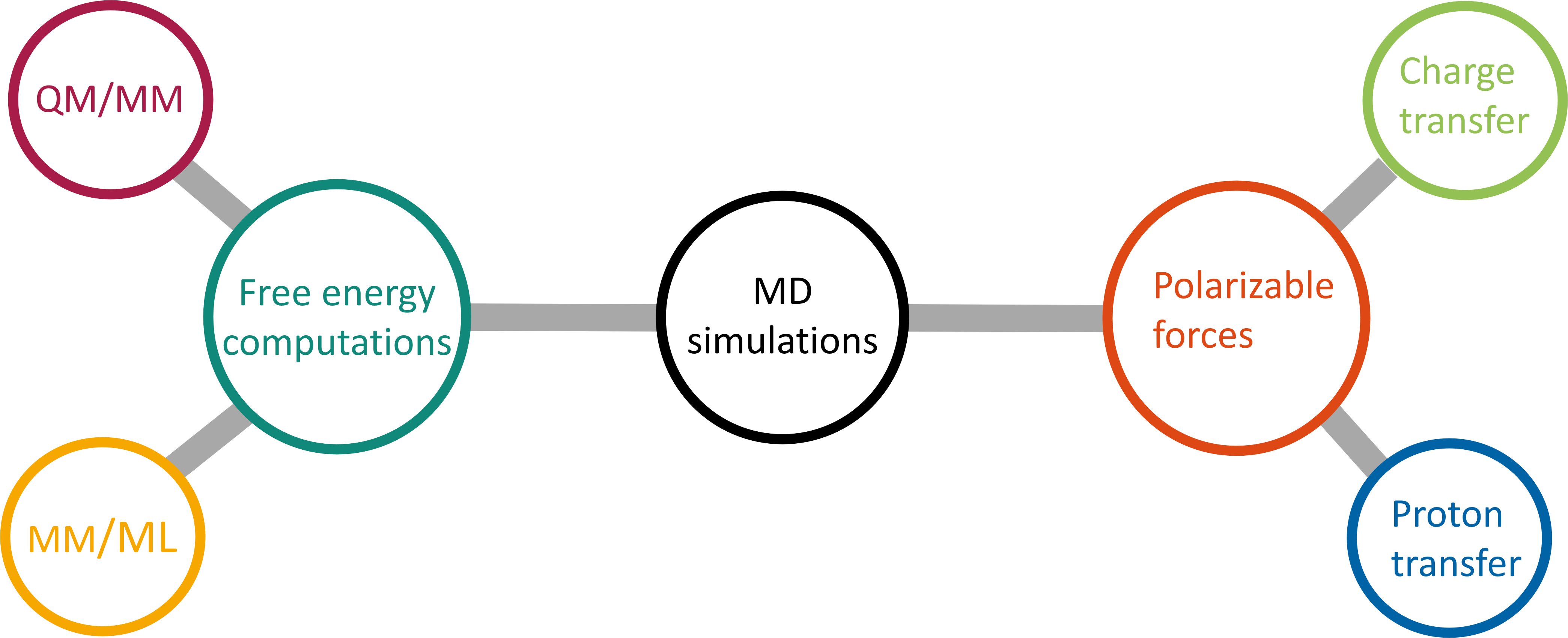
We are experts in molecular dynamics simulations, with a particular emphasis on advanced free energy calculations and the
incorporation of polarizable force fields. Our proficiency in free energy calculations also includes
quantum mechanics/molecular mechanics (QM/MM) and molecular mechanics/machine learning (MM/ML) hybrid approaches.
Polarizable forces are essential to simulate systems containing large amounts of charged species.
Furthermore, they enable to model charge and proton transfer.
For both, simulation and analysis, we develop code for (highly parallel) computing on
modern CPU and GPU architecture.
Free energy calculations
The change in free energy ΔA =
A(β)-A(α) between two states α, β provides the
single criterion for the spontaneity of a chemical or biological
process. Computer simulations can not only determine free energy
differences of interest, but they also make possible a microscopic
(atomistic) explanation of the result obtained. Research interests concern both
methodology, as well as application.
FWF P19100: "Towards more accurate and efficient free energy simulations"
Grant holder
S. Boresch
Funding period
09/2006 - 08/2010
Computational spectroscopy
To make spectroscopic calculations on nuclear motion feasible
our molecular dynamics simulations usually are atom-resolved and based on classical mechanics.
According to the requirements also hybrid (quantum mechanical), polarizable,
coarse-grained, or multi-scale models are designed and implemented.
Molecular dynamics simulations offer a powerful computational microscope that allows us to observe and analyze the
dynamic behavior of biomolecules. They provide insights into the structural, functional, and thermodynamic properties
of biological systems. In addition to classical protein-ligand binding of medical relevant proteins in cancer research,
we are interested in ion channels, integral membrane proteins that facilitate the selective transport of ions across cell membranes,
are of paramount interest due to their critical roles in cellular signaling, homeostasis, and physiology.
By leveraging the capabilities of MD simulations, we aim to bridge the gap between theoretical models and experimental
observations, offering a comprehensive view of biomolecular dynamics. Our approach encompasses a wide range of methodologies,
including classical MD simulations, free energy calculations, and the integration of polarizable force fields including proton transfer,
to accurately capture the essence of biomolecular interactions.
ASEA UNINET ASEA 51/2024: Exploring pH dependent structural dynamics in urinary HSA fragments for CKD detection
Grant holder
C. Schröder / P. Ponprayoon
Funding period
01/2025 - 12/2025
ASEA UNINET ASEA 50/2024: Polarizable molecular dynamics simulations of the proton transport in the influenza M2 ion channel
As a simple definition given by Paul Walden in 1914, ionic liquids are commonly recognized as salts with a melting point
below 100° C. Popular cations are imidazoliums but other organic heterocyclic cations such as pyridinium or pyrrolidinium are also possible.
In addition, ammonium, phosphonium and sulfonium cations with linear, branched or functionalized side chains have been used.
Typical anorganic anions comprise halides, alkylsulfates, alkylsulfonates and in particular bis(trifluoromethyl-sulfonyl)imide.
The plethora of cation/anion combinations allows for variation of the physico-chemical properties over a very broad range and
can be further fine-tuned by side chain modifications of both, cations and anions.

 We explore the chemical world by modeling large molecular systems on the computer
and looking at their diverse properties such as structure, energies, dynamics and spectroscopic quantities.
We explore the chemical world by modeling large molecular systems on the computer
and looking at their diverse properties such as structure, energies, dynamics and spectroscopic quantities.