
Towards prediction of electrostatic parameters for force fields that explicitly treat electronic polarization
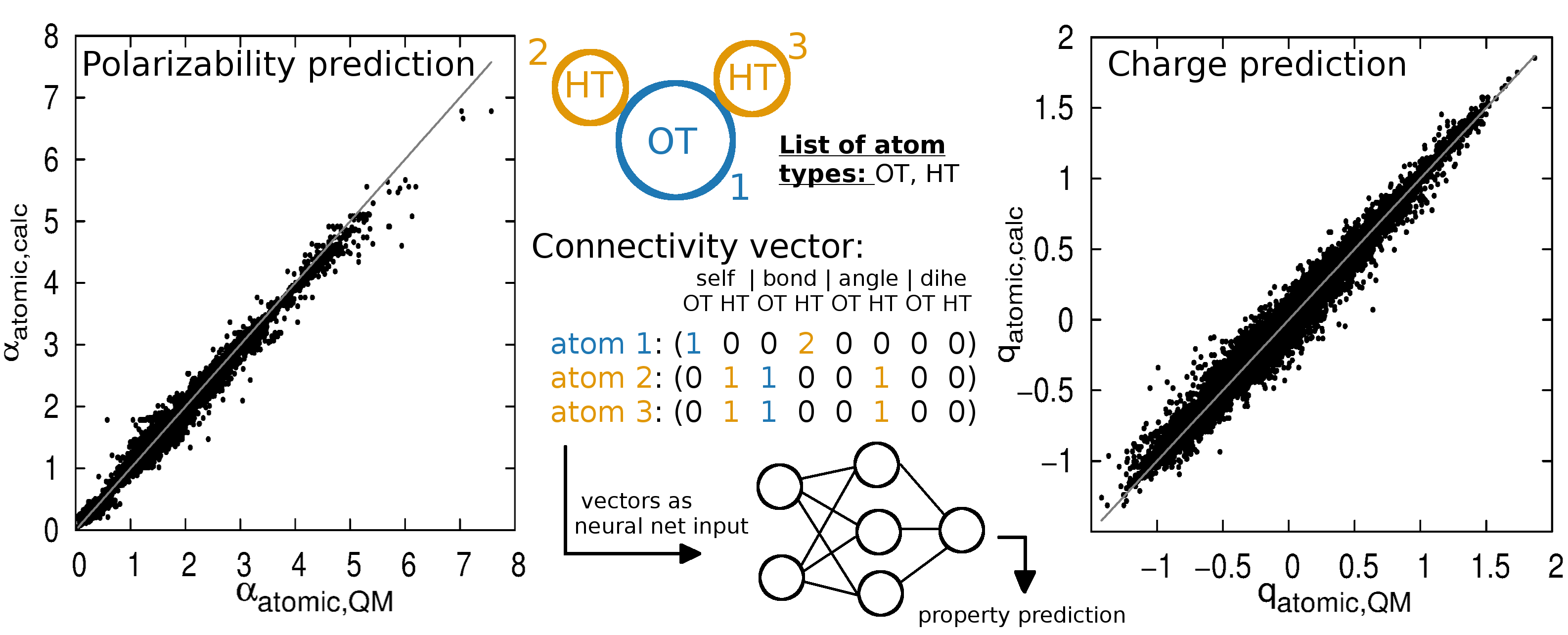 The derivation of atomic polarizabilities for polarizable force fields has been a long-standing problem. These polarizabilities were often
refined manually rendering an automated assignment difficult and hampering reproducibility and transferability of the obtained values.
In a recent manuscript, we developed a
python based tool to estimate partial charges and atomic polarizabilities
for force fields using the atom types of CGENFF. Please follow the link for
downloading the corresponding code from Alex MacKerells homepage.
The derivation of atomic polarizabilities for polarizable force fields has been a long-standing problem. These polarizabilities were often
refined manually rendering an automated assignment difficult and hampering reproducibility and transferability of the obtained values.
In a recent manuscript, we developed a
python based tool to estimate partial charges and atomic polarizabilities
for force fields using the atom types of CGENFF. Please follow the link for
downloading the corresponding code from Alex MacKerells homepage.The predictor script allows you to calculate approximate atomic polarizabilities and partial atomic charges, as well as lonepairs and anisotropic polarizabilities for organic compounds. You need to supply a mol2 or a CGenFF stream file that contains the CGenFF atom types and atom connectivity. Running in the mol2 mode requires that the CGenFF program be installed on your system. Check the file setup/settings.dat to specify your executable names of CGenFF (if needed), and the details of the prediction algorithms (using linear increments or a neural net, calculate lonepairs, verbosity).
Please note that currently only the geometry of lone pairs is set up. Please redistribute partial charges between the atom and the corrresponding lone pairs. This feature will be added in future version.
Note that CGenFF stream files may be obtained for mol2 files using the online CGenFF server at https://cgenff.umaryland.edu/
Solved Bugfixes:
| 2019/03/26 | Fixed bug in manual charge mode |