
Computational Methods
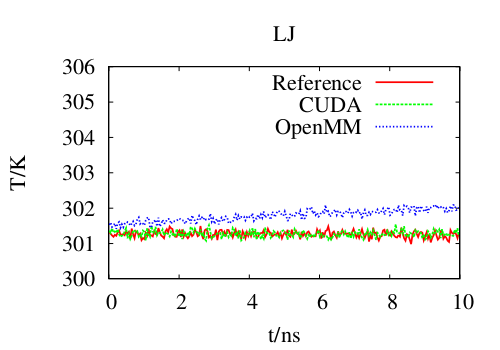
We are actively participating in the development of the CHARMM simulation program, with present emphasis on utilizing the GPU to accelerate simulations. The goal is acceptable performance while retaining the accuracy and precision of the double precision CPU code. The figure on the left compares temperature conservation of regular CHARMM (red), our GPU code (green) and CHARMM using the OpenMM library (blue), with OpenMM exhibiting a problematic rise in temperature even over 10~ns.
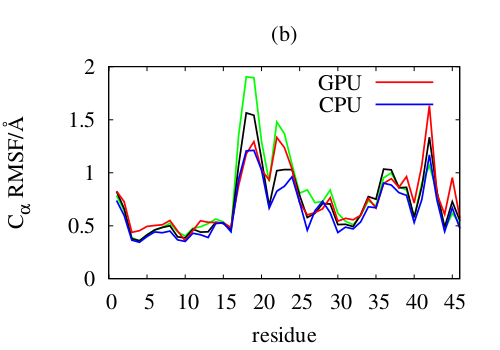
Further, the code is fully compatible with the polarizable and coarse-grained force fields under development. The figure on the right shows the Cα root mean square fluctuation of crambin simulated with an experimental version of the polarizable protein force field (CPU: blue; GPU: green, red, black). One GPU easily outperforms regular CHARM on 16 CPU cores, in most cases even on 32 cores.
For our analysis, trajectory data of several dozens of nanoseconds have to be postprocessed and then used to compute physical properties of interest. Consequently, the corresponding tools should also be parallized to handle such amounts of data in a reasonable time.