
Free energy calculations
The change in free energy ΔA = A(β)-A(α) between two states α, β provides the single criterion for the spontaneity of a chemical or biological process. Computer simulations can not only determine free energy differences of interest, but they also make possible a microscopic (atomistic) explanation of the result obtained. Research interests concern both (1) methodology, as well as (2) application.
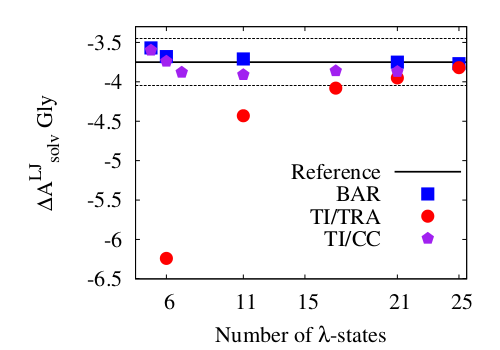 (1) E.g., the plot on the left shows how the number of required
intermediate states can be reduced by choosing either Bennett's
acceptance ratio method or an appropriate method for numerical
quadrature in thermodynamic integration.
Based on results, we are currently looking into ways to
increase the computational efficiency of free energy simulations based
on hybrid quantum mechanical/molecular mechanical calculations
(in collaboration with USF and NIH).
(1) E.g., the plot on the left shows how the number of required
intermediate states can be reduced by choosing either Bennett's
acceptance ratio method or an appropriate method for numerical
quadrature in thermodynamic integration.
Based on results, we are currently looking into ways to
increase the computational efficiency of free energy simulations based
on hybrid quantum mechanical/molecular mechanical calculations
(in collaboration with USF and NIH).
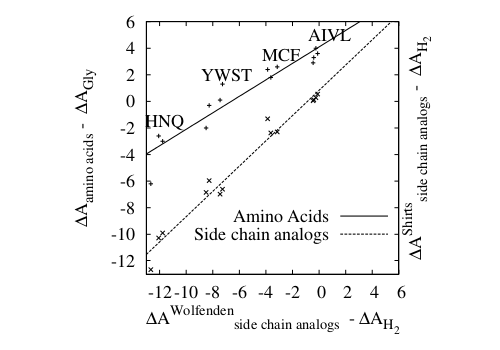 (2) The figure on the right demonstrates that the affinity of amino
acids for solvent water (solid line) differs markedly from that of
their side chain analogs (dotted line) as can be seen from the
significant lower slope. The reasons for this are self-solvation (SS)
and solvent-exclusion (SE). While standard models to rationalize the
solvent affinity of amino acids take SE into account, SS has been
almost completely overlooked so far. This finding puts into question
some common assumptions concerning the role of water (i.e., solvation)
for the stability of proteins.
(2) The figure on the right demonstrates that the affinity of amino
acids for solvent water (solid line) differs markedly from that of
their side chain analogs (dotted line) as can be seen from the
significant lower slope. The reasons for this are self-solvation (SS)
and solvent-exclusion (SE). While standard models to rationalize the
solvent affinity of amino acids take SE into account, SS has been
almost completely overlooked so far. This finding puts into question
some common assumptions concerning the role of water (i.e., solvation)
for the stability of proteins.